

联系方式【微信:hjjk0006】【微信:hjjk1002】
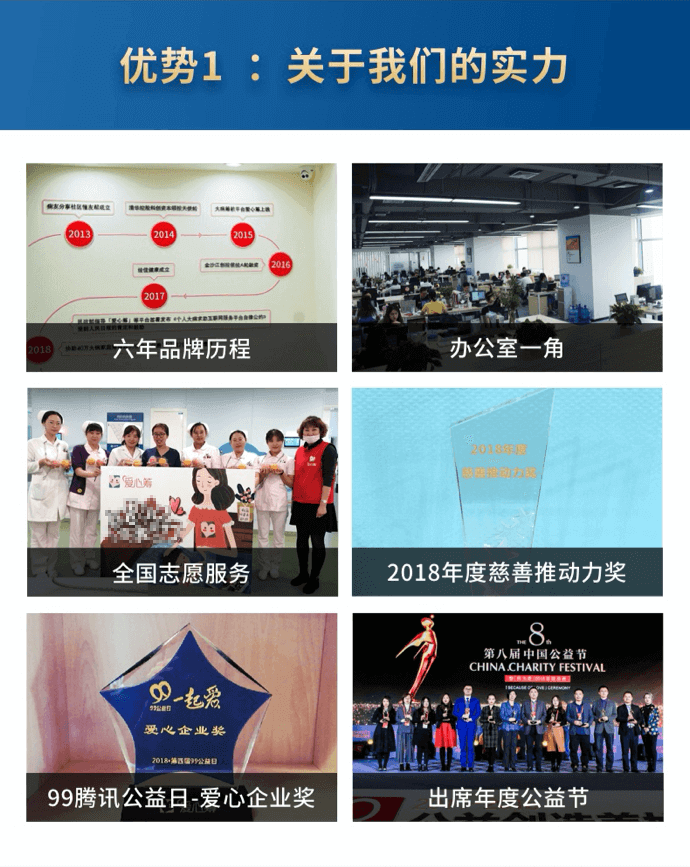
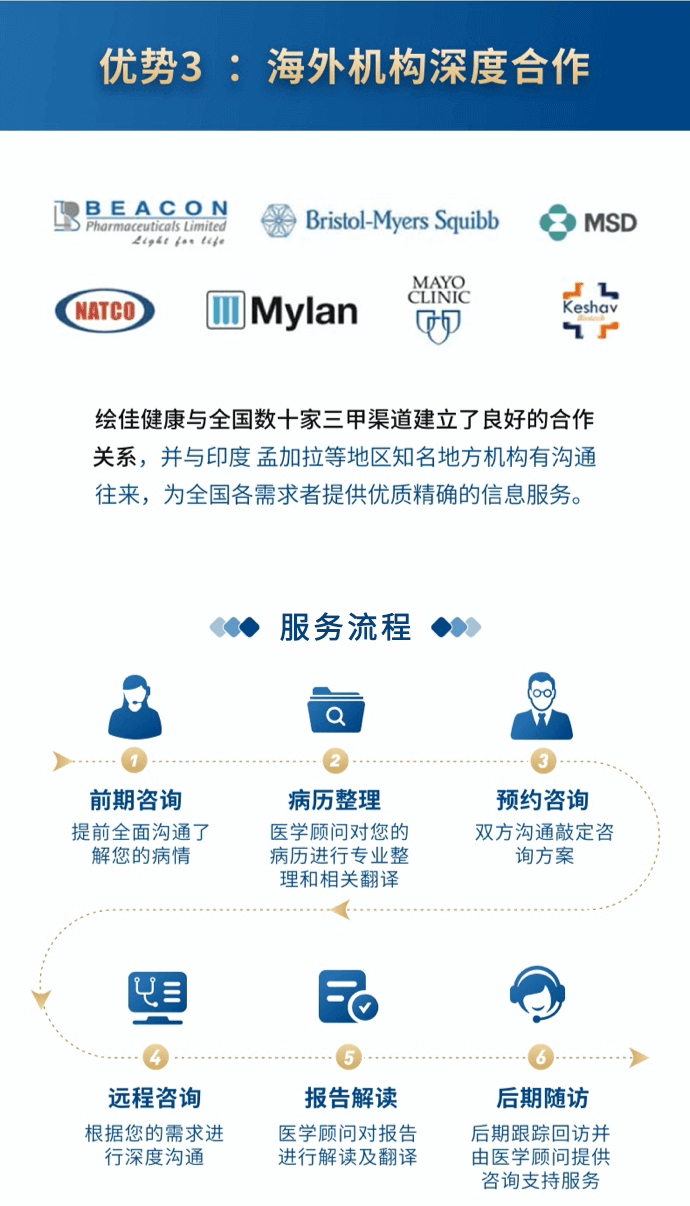
绘佳医疗是全球寻药和跨境医疗服务的领导者,为大病患者提供了出国看病、药房对接、病历翻译、远程咨询等服务,致力于帮助大病家庭改善医疗资金的支付能力和帮助患者降低治疗花销。
 免责声明:本文所表达的任何关于疾病的建议都不应该被视为医生的建议或替代品,请咨询您的治疗医生了解更多细节。本站信息仅供参考,绘佳医生不承担任何责任。
免责声明:本文所表达的任何关于疾病的建议都不应该被视为医生的建议或替代品,请咨询您的治疗医生了解更多细节。本站信息仅供参考,绘佳医生不承担任何责任。




热门评论