

在欧美临床与药品监管体系中,洛拉替尼(lorlatinib)原研药的临床疗效与仿制药的质量评估是一个严肃而专业的话题。洛拉替尼作为第三代间变性淋巴瘤激酶(ALK)抑制剂,其原研产品在美国食品药品监督管理局(FDA)与欧洲药品管理局(EMA)均获批准,用于ALK阳性晚期非小细胞肺癌(NSCLC)的治疗,这一批准基于CROWN III期临床试验的强有力数据支持(例如在欧洲批准文件中指出其显著改善无进展生存)FDA及EMA对原研药的批准具有严格的药效与安全性评估,其公开资料显示洛拉替尼在多国获批用于一线及既往治疗后继续治疗的适应症
关于仿制药“哪个好”,目前在美国与欧洲与老挝尚无大量来自《新英格兰医学杂志》《柳叶刀肿瘤学》等权威期刊的独立对比数据。欧美仿制药的开发必须遵循严格的生物等效性标准:FDA与EMA要求仿制药在吸收(AUC)和最高血药浓度(Cmax)的90%置信区间落在80%–125%范围内,才能被认为在疗效和安全性上与原研药等效
这一规定体现了欧美对仿制药质量严格的科学要求,但并不等于所有仿制药在临床表现上一定完全一致:如同生物等效性领域的研究所指出,不同仿制产品在复杂药物体系中可能存在差异,这需要具体药品和临床数据去验证
因此,在当前公开、权威的数据中,老挝第二的科学证据支持某一洛拉替尼仿制药优于另一种。在评估仿制药时,而不是简单的“哪个好”比较。点击咨询
联系方式【微信:hjjk1002】【微信:hjjk0006】
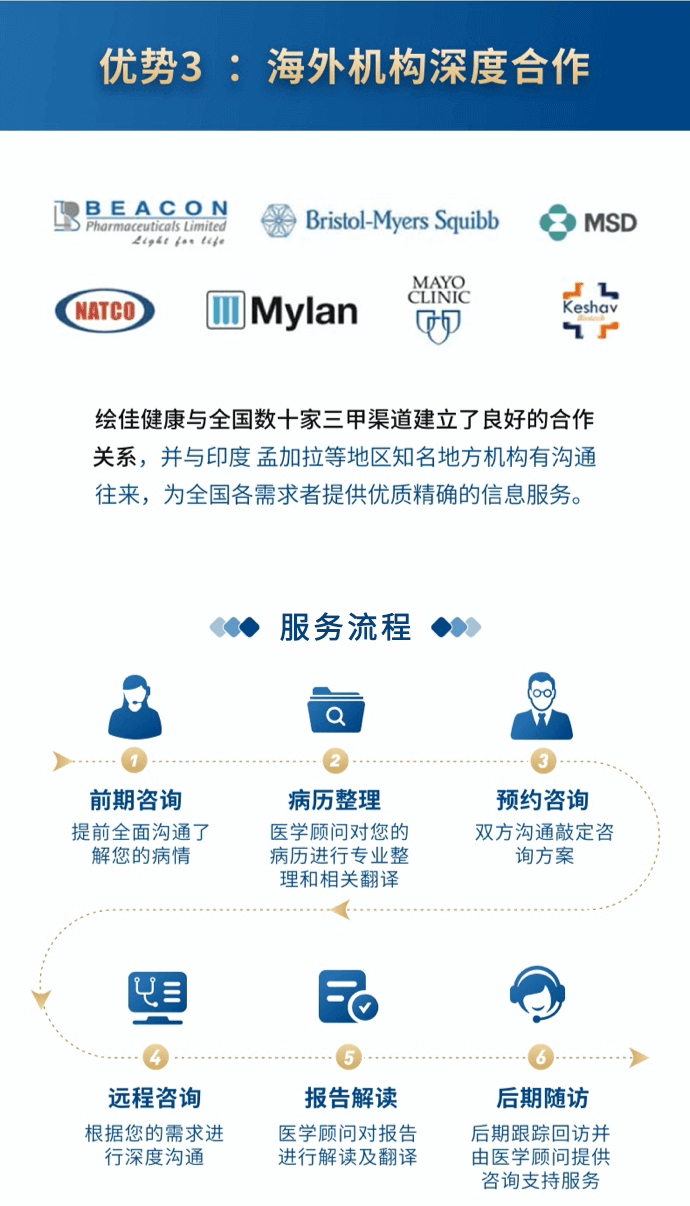
绘佳医疗是全球寻药和跨境医疗服务的领导者,为大病患者提供了出国看病、药房对接、病历翻译、远程咨询等服务,致力于帮助大病家庭改善医疗资金的支付能力和帮助患者降低治疗花销。
 免责声明:本文所表达的任何关于疾病的建议都不应该被视为医生的建议或替代品,请咨询您的治疗医生了解更多细节。本站信息仅供参考,绘佳医生不承担任何责任。
免责声明:本文所表达的任何关于疾病的建议都不应该被视为医生的建议或替代品,请咨询您的治疗医生了解更多细节。本站信息仅供参考,绘佳医生不承担任何责任。




热门评论